1.理論模型
1.1 史密斯政策執(zhí)行過程的分析框架
20 世紀(jì)70 年代�,美國學(xué)者史密斯在《政策執(zhí)行過程》一文中����,首次提出分析政策執(zhí)行過程及其影響因素的理論模型���,該模型稱為“史密斯模型”�����,又稱“史密斯政策執(zhí)行過程模型”����。史密斯認(rèn)為,政策執(zhí)行受多方因素影響�,主要體現(xiàn)在理想化政策、執(zhí)行機(jī)構(gòu)����、目標(biāo)群體、政策環(huán)境等4 個變量�,這4 個變量緊密相關(guān),彼此互相作用與影響�。史密斯模型通過分析上述4 個變量之間的關(guān)系,使政策分析的脈絡(luò)更加清晰��,能用于更準(zhǔn)確地分析和解決政策執(zhí)行過程中的問題�����。首先����,理想化政策是指在毫無干擾的環(huán)境下達(dá)到的完美狀態(tài)��,現(xiàn)實(shí)中難以實(shí)現(xiàn)����,這是政策執(zhí)行前最重要的一環(huán)�。其次,執(zhí)行機(jī)構(gòu)是指政府中具體負(fù)責(zé)政策執(zhí)行的機(jī)構(gòu)���,涉及執(zhí)行人員的情況以及領(lǐng)導(dǎo)者的領(lǐng)導(dǎo)模式���、技巧等。再次���,目標(biāo)群體是指政策執(zhí)行的實(shí)施對象�����,即受政策影響最大的群體�����。最后,政策環(huán)境是指與政策執(zhí)行有關(guān)聯(lián)的影響因子,包括政治���、經(jīng)濟(jì)�����、社會�����、文化因素等����。
1.2 藥品上市后安全性研究政策執(zhí)行分析框架
史密斯認(rèn)為����,政策執(zhí)行過程是上述4 種變量之間互相作用、互相協(xié)調(diào)的過程�。在此過程中,無論是政策參與者還是執(zhí)行者�����,都會受到過程變量的影響���,這兩者之間的政策張力以及因此帶來的沖突可能會引發(fā)“緊張”的政策關(guān)系���。針對這種“緊張”的政策關(guān)系����,可利用制度化或非制度化的措施處理出現(xiàn)的過程性問題并進(jìn)行問題反饋�,通過協(xié)調(diào)與修正變量之間的關(guān)系,使政策得以有效執(zhí)行���?��;谑访芩鼓P椭械? 個影響因子,構(gòu)建PASS 政策執(zhí)行分析框架���,如圖1 所示�����,這4 個影響因子會對PASS 政策執(zhí)行產(chǎn)生一定的影響���。

2.藥品上市后安全性研究政策執(zhí)行影響因素分析
2.1 理想化政策
2019 年新修訂《藥品管理法》實(shí)施后,MAH 制度開始在全國范圍實(shí)施�����。MAH 制度實(shí)施前,藥品上市后階段涉及藥品流通質(zhì)量管理��、上市后監(jiān)測�、風(fēng)險管理����、批準(zhǔn)后研究等,其責(zé)任主體包括藥品生產(chǎn)企業(yè)�、經(jīng)營企業(yè)、醫(yī)療機(jī)構(gòu)等��,但未規(guī)定研發(fā)者對上市后藥品的義務(wù)和責(zé)任��。MAH 制度實(shí)施后����,藥品上市后階段的責(zé)任主體為MAH。根據(jù)《藥品管理法》第七十七條����,MAH 應(yīng)當(dāng)制定藥品上市后風(fēng)險管理計劃,主動開展藥品上市后研究�����,對藥品的安全性、有效性和質(zhì)量可控性進(jìn)行進(jìn)一步確證�,加強(qiáng)對已上市藥品的持續(xù)管理[4]。2020 年新修訂《藥品注冊管理辦法》進(jìn)一步細(xì)化了MAH 制度的框架�����、適用范圍等���。MAH 制度的實(shí)施��,降低了藥品研發(fā)機(jī)構(gòu)的成本投入���,激發(fā)了藥品研發(fā)積極性。然而�����,MAH 主動收集不良反應(yīng)并報告的意識仍有待提高�,作為藥品安全“第一責(zé)任人”,部分MAH 未貫徹落實(shí)《藥品管理法》相關(guān)規(guī)定�。
GVP 是我國建立藥物警戒制度以來出臺的第一個藥物警戒規(guī)范性文件, 論述了PASS 相關(guān)內(nèi)容����。但與美國���、日本等國家相比, 我國對于PASS 還沒有詳細(xì)的規(guī)定�。《藥品管理法》第一百三十四條規(guī)定��,MAH未按照規(guī)定開展藥品不良反應(yīng)(adverse drug reaction���,ADR)監(jiān)測或者報告疑似ADR的,責(zé)令限期改正�,給予警告;逾期不改正的����,責(zé)令停產(chǎn)停業(yè)整頓, 并處十萬元以上一百萬元以下的罰款��?�?梢?���,目前我國對MAH 違法行為的處罰措施限于警告和罰款��, 對MAH 的激勵��、警示和威懾作用仍有待加強(qiáng)��。
2.2 執(zhí)行機(jī)構(gòu)
PASS 政策的執(zhí)行機(jī)構(gòu)主要是國家藥監(jiān)局��、省級藥品監(jiān)管部門以及衛(wèi)生行政部門等�。藥品監(jiān)管部門和衛(wèi)生行政部門的工作職責(zé)包括保障公眾用藥安全�,并對ADR 監(jiān)測和報告進(jìn)行監(jiān)督和管理。藥物警戒貫穿藥物發(fā)展全過程��,包括上市前和上市后階段�,其中上市后監(jiān)管包括ADR 監(jiān)測和報告。MAH 制度實(shí)施前����,我國藥物警戒工作存在一些不足,如相關(guān)法律法規(guī)不完善����、信息系統(tǒng)不成熟、藥物警戒體系不健全等����,ADR 報告和監(jiān)測以被動為主��,對于新的���、嚴(yán)重的ADR 分析能力有限,藥物警戒相關(guān)法律法規(guī)制定刻不容緩����;MAH 制度的實(shí)施為建立健全藥物警戒相關(guān)法律法規(guī)提供了依據(jù),促進(jìn)了我國GVP 的出臺����,推動我國藥物警戒發(fā)展進(jìn)入新階段��。同時�����,MAH 在ADR 監(jiān)測和報告方面的主動性也有所提高����,但其主動性意識仍需進(jìn)一步加強(qiáng)。
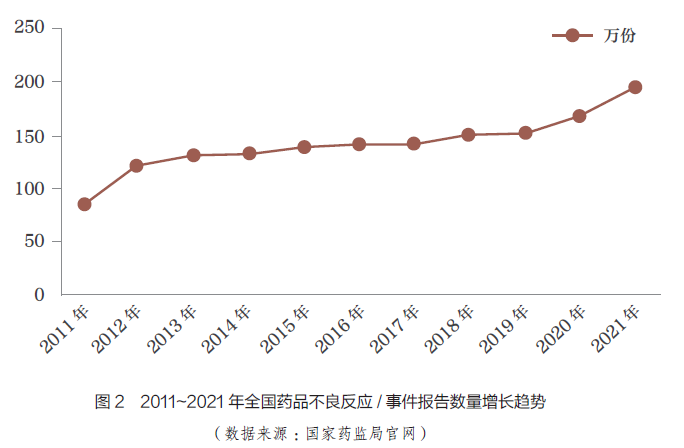
隨著MAH���、醫(yī)療機(jī)構(gòu)以及患者對ADR 的重視�����,ADR 事件報告的數(shù)量逐年增加����。藥品不良事件是指在藥物治療過程中發(fā)生的任何不幸的醫(yī)療衛(wèi)生事件,而這種事件不一定與藥物治療存在因果關(guān)系[9]����。根據(jù)國家藥監(jiān)局統(tǒng)計,1999~2021 年�����, 全國ADR監(jiān)測網(wǎng)絡(luò)累計收到《藥品不良反應(yīng)/ 事件報告表》1883 萬份,且每年ADR 事件數(shù)量不減反增,2011~2021 年全國不良反應(yīng)/ 事件報告數(shù)量增長趨勢如圖2 所示��。2021 年,全國不良反應(yīng)/ 事件共196.2 萬份����,較2020 年增長17.1%,但來源于MAH 的報告數(shù)量僅占同期總數(shù)的4.1%�����,較2020 年的3.9% 并沒有明顯增長。由此可見����,我國對ADR 的監(jiān)測亟需進(jìn)一步加強(qiáng)。2013~2021年����,國家藥監(jiān)局發(fā)布的國家ADR監(jiān)測年度報告來源情況如圖3 所示。數(shù)據(jù)顯示�,近10 年來,醫(yī)療機(jī)構(gòu)報告的ADR 事件占大多數(shù)����,其次是藥品生產(chǎn)經(jīng)營企業(yè);近3年來���,MAH 報告的ADR 數(shù)量雖有所增加,但增幅不大����,報告數(shù)量所占比例仍較低,我國GVP制度的落地還需要不斷完善相關(guān)配套制度和規(guī)范��。
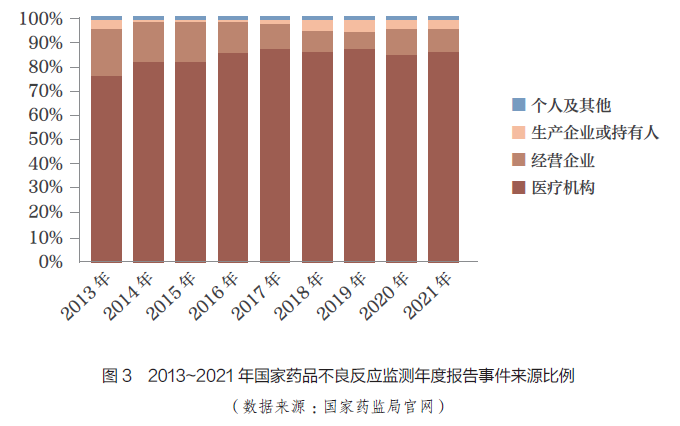
在MAH 制度實(shí)施前,ADR報告的責(zé)任主體是藥品生產(chǎn)和經(jīng)營企業(yè)�、醫(yī)療機(jī)構(gòu)、個人及其他��,但從ADR 報告的數(shù)量來看���,藥品生產(chǎn)和經(jīng)營企業(yè)報告數(shù)量較少���,主要原因在于企業(yè)追求利益最大化,不愿在ADR 監(jiān)測和報告中投入更多人力����、物力和財力,部分企業(yè)認(rèn)為公眾知曉ADR 報告會影響產(chǎn)品銷量和企業(yè)形象���。相信隨著MAH 制度的持續(xù)推進(jìn)��,上述狀況將得到改善�����。
2.3 目標(biāo)群體
目標(biāo)群體是指政策執(zhí)行的實(shí)施對象���。PASS 實(shí)施對象包括MAH�����、醫(yī)療機(jī)構(gòu)及藥品生產(chǎn)經(jīng)營企業(yè)等���。郭莎莎等 認(rèn)為,河北省僅有少部分MAH 開展風(fēng)險評估�,原因可能是MAH 把更多的資源和精力投入到藥品上市前研究階段,而減少了藥品上市后風(fēng)險管控的投入�����。方樂敏等認(rèn)為���,上海市部分MAH 存在未對藥品安全數(shù)據(jù)進(jìn)行回顧性評價�����、未實(shí)施評價和控制等問題����,即使有些MAH 發(fā)現(xiàn)藥品存在風(fēng)險信號��,也沒有進(jìn)行控制�����,無法實(shí)現(xiàn)風(fēng)險管理的閉環(huán)�����。目前���,我國ADR 報告來源以醫(yī)療機(jī)構(gòu)為主����,藥品生產(chǎn)經(jīng)營企業(yè)為輔�����,存在收集渠道不健全��、漏報瞞報����、信息利用率低、主動監(jiān)測意識不足等問題�����。而藥物警戒制度的核心內(nèi)容是藥品風(fēng)險管理,MAH 需要探測藥品全生命周期中的隱患信號�,在未發(fā)生嚴(yán)重ADR 事件時就加以控制,確保藥品上市前與上市后的安全性�����。
藥品生產(chǎn)企業(yè)是保障藥品安全的責(zé)任主體��。部分藥品生產(chǎn)企業(yè)對藥品上市前安全性研究的投入比上市后多�����,認(rèn)為藥品上市后安全性研究投入會增加企業(yè)負(fù)擔(dān)���。企業(yè)加大對藥物警戒的資金投入�����,可能不會給企業(yè)帶來直接的經(jīng)濟(jì)利益��, 但可以保障企業(yè)獲得更大的經(jīng)濟(jì)利益��。藥品生產(chǎn)企業(yè)有必要對上市后的藥品進(jìn)行監(jiān)測����, 降低藥品上市后的安全性風(fēng)險����。
2.4 政策環(huán)境
我國藥品監(jiān)管由國家藥監(jiān)局、省級藥監(jiān)局����、市縣藥監(jiān)部門三級管理。由于各省份�、城鄉(xiāng)之間醫(yī)療水平不同、資源分布不均�、監(jiān)管能力存在差異等,加之跨省監(jiān)管協(xié)作機(jī)制仍有待完善��,MAH制度順利實(shí)施的難度較大����。目前,我國藥品監(jiān)管部門對藥品的審批更加嚴(yán)格����,并加強(qiáng)了上市后監(jiān)管。然而����,每年ADR 報告數(shù)量仍在增加���,原因之一是ADR安全性評價機(jī)制仍有待完善。少數(shù)醫(yī)療機(jī)構(gòu)為了通過審批偽造材料��, 加之審批時間縮減����, 導(dǎo)致藥品審批監(jiān)管難度加大, 藥品質(zhì)量和安全性受到影響��。新藥上市前安全性研究需要經(jīng)過臨床試驗(yàn)階段�����,試驗(yàn)會受到時間���、用藥人群等影響�����,導(dǎo)致新藥上市后的風(fēng)險不能被準(zhǔn)確估量�����;而隨著上市后用藥人群的擴(kuò)大���,風(fēng)險隱患也在增加����。因此��,相關(guān)部門有必要開展PASS 研究����。
隨著人們生活質(zhì)量的提高��,醫(yī)療衛(wèi)生需求日益增加���, 國家出臺了一系列保證藥品質(zhì)量和安全的政策���, 同時加深了人們對藥品安全的認(rèn)識。GVP 強(qiáng)調(diào)了在開展PASS 過程中的風(fēng)險評估和控制���,如MAH 應(yīng)當(dāng)監(jiān)測PASS 期間的安全性信息����, 發(fā)現(xiàn)任何可能影響藥品獲益- 風(fēng)險平衡的新信息, 應(yīng)當(dāng)及時開展評估����,并采取適宜風(fēng)險控制措施;研究中發(fā)現(xiàn)可能嚴(yán)重危害患者生命安全或公眾健康的藥品安全問題時��,MAH 應(yīng)當(dāng)立即采取暫停銷售和使用�����、召回等緊急控制措施�����,并將召回和處理情況向省級藥品監(jiān)管部門和ADR 監(jiān)測機(jī)構(gòu)報告�。為了確保受試者或用藥者的安全,MAH 要在整個PASS 過程中貫徹風(fēng)險管理理念��, 并對藥品全生命周期進(jìn)行監(jiān)測�。
3.藥品上市后安全性研究政策執(zhí)行建議
3.1 完善PASS 政策,出臺PASS 管理規(guī)范和條例
我國的PASS 法律體系是以法律為主��、法規(guī)為輔����,共同指導(dǎo)MAH 進(jìn)行PASS�,同時由相關(guān)部門指導(dǎo)和監(jiān)督研究全過程�。MAH 應(yīng)建立藥品安全體系,對藥品上市后安全進(jìn)行監(jiān)測�,包括ADR 監(jiān)測與報告流程規(guī)范、藥品上市前后風(fēng)險管理相關(guān)文件�����、藥品上市后再評價相關(guān)規(guī)定等�。MAH 制度的不斷完善����,有利于MAH 開展PASS。
如上所述�����, 目前我國在PASS 政策執(zhí)行過程中��,對違法行為的行政處罰方式僅是警告和罰款��。為保證PASS 政策的實(shí)施����,建議在藥品監(jiān)管相關(guān)法律法規(guī)中對PASS 作出更詳細(xì)的規(guī)定,對違法行為制定更嚴(yán)厲的處罰措施(如停止銷售相關(guān)產(chǎn)品、吊銷《藥品生產(chǎn)許可證》等)����,以進(jìn)一步督促M(fèi)AH 自覺遵守PASS 相關(guān)規(guī)定。同時�����,建議相關(guān)部門出臺PASS 相關(guān)管理規(guī)范等�����,從而更好指導(dǎo)MAH 開展PASS�����。
3.2 加強(qiáng)MAH 責(zé)任落實(shí)及對PASS 的監(jiān)管�����,加大PASS 投入
MAH 是藥物警戒的責(zé)任主體�����,職責(zé)包括對ADR 進(jìn)行監(jiān)測和報告���。從已發(fā)生的ADR 事件來看��,開展ADR 監(jiān)測不僅有助于發(fā)現(xiàn)上市前受環(huán)境等因素影響未被發(fā)現(xiàn)的安全性問題����,還有助于發(fā)現(xiàn)產(chǎn)品在流通或用藥過程中出現(xiàn)的問題。同時�,ADR 監(jiān)測和報告的結(jié)果可作為藥品安全性評價的依據(jù),從而保障藥品質(zhì)量和公眾用藥安全��。此外����,藥品生產(chǎn)經(jīng)營企業(yè)在開展ADR 監(jiān)測時��,還可向公眾展示最新的產(chǎn)品信息��,這種具有主動性�、自覺性的做法有利于企業(yè)樹立良好形象,增加公眾對企業(yè)的信任�����。因此����,從長遠(yuǎn)來看�����,加大對PASS 的投入有利于企業(yè)發(fā)展��。
3.3 提高藥品上市后的風(fēng)險管理
在PASS 政策執(zhí)行過程中��,需要進(jìn)行藥品上市后風(fēng)險管理���,包括識別藥品風(fēng)險信號、啟動風(fēng)險管理計劃�、藥品安全性再評價、風(fēng)險效益再評價等��。MAH 實(shí)施藥物警戒制度時需改變思想觀念��,由被動轉(zhuǎn)變?yōu)橹鲃颖O(jiān)測�����、識別�����、評估與控制藥品風(fēng)險。在監(jiān)測階段�����,MAH 應(yīng)主動收集藥物警戒相關(guān)信息并按要求上報����,完善ADR 網(wǎng)絡(luò)信息系統(tǒng)和藥品監(jiān)測機(jī)制,全面監(jiān)測藥品風(fēng)險��。在識別風(fēng)險階段�����,MAH 應(yīng)增強(qiáng)對監(jiān)測信息的分析能力�,識別信息中是否存在潛在風(fēng)險信號。在評估階段���,MAH 應(yīng)重點(diǎn)關(guān)注具有潛在風(fēng)險的信息,研究風(fēng)險發(fā)生機(jī)制���,定期對藥品開展風(fēng)險評估���,包括藥品安全性評價�����、藥品潛在風(fēng)險識別��、主動開展PASS 等��。在控制階段���,MAH 應(yīng)根據(jù)風(fēng)險評估結(jié)果,對存在的風(fēng)險采取相應(yīng)的解決措施�,并撰寫年度ADR 報告,減少藥品對公眾健康的危害�����。
3.4 健全藥品上市后安全性評價體系
ADR 數(shù)據(jù)信息是開展藥物警戒工作的基礎(chǔ)����。2016 年,原國家食品藥品監(jiān)督管理總局藥品評價中心開發(fā)了中國醫(yī)院藥物警戒系統(tǒng)(CHPS)�����,通過該系統(tǒng)可主動監(jiān)測是否存在ADR 風(fēng)險���。該監(jiān)測手段靈敏度高����、便于數(shù)據(jù)分析,優(yōu)化了醫(yī)療機(jī)構(gòu)上報ADR 事件的流程����,提高了醫(yī)療機(jī)構(gòu)對ADR的監(jiān)測水平,為藥品上市后安全性評價提供了數(shù)據(jù)信息支撐��。
藥品上市后安全性評價體系的構(gòu)建和完善是PASS 的內(nèi)容之一��,對于開展PASS 具有重要意義��。藥品上市前和上市后都需要進(jìn)行風(fēng)險評價����,上市后安全性評價有利于識別和確認(rèn)藥品上市后是否存在安全隱患,如存在安全隱患����,則需要采取暫停銷售、召回藥品等措施�����,從而有效防止更嚴(yán)重的ADR 事件發(fā)生�。在藥品審批階段,建議藥品監(jiān)管部門加強(qiáng)對申請資料的審查��,確保資料真實(shí)可靠����。為保證安全性評價的真實(shí)性和準(zhǔn)確性,建議選擇德才兼?zhèn)?���、?jīng)驗(yàn)豐富的醫(yī)師作為藥品安全性評價專家。此外����,建議藥品監(jiān)管部門完善新藥上市后安全性評價體系,定期對藥品開展抽檢和ADR 監(jiān)測���。
近年來���,我國不斷出臺規(guī)范藥物警戒相關(guān)文件,藥物警戒體系逐漸完善�,相關(guān)法律法規(guī)不斷成熟,為MAH 開展PASS 提供了保障。MAH 制度雖已被寫入《藥品管理法》中����,但仍處于推進(jìn)階段,還需要不斷完善���。GVP 的頒布�����,促進(jìn)了MAH 制度和PASS制度的推行����,但同時需要注意���,ADR 監(jiān)測和管理方面還需完善��,需要出臺PASS 管理規(guī)范等來進(jìn)一步明確PASS 相關(guān)內(nèi)容���,加強(qiáng)對PASS 的監(jiān)管和風(fēng)險管理,建立健全藥品上市后安全性評價體系�,從而最大程度地降低藥品安全風(fēng)險,保障公眾生命安全與健康�����。
(來源:中國醫(yī)藥報)