近年來(lái),細(xì)胞與基因療法(Cell and Gene Therapy, CGT)已逐漸成為最引人注目的領(lǐng)域之一��,在帶來(lái)顛覆性治療的同時(shí),CGT市場(chǎng)也迎來(lái)蓬勃發(fā)展���。2016-2020年��,全球CGT市場(chǎng)規(guī)模年復(fù)合增長(zhǎng)率達(dá) 153.3%�����,2020年為20.75億美元�。CGT 2022年全球市場(chǎng)體量將達(dá) 到200億美元��,保持50%的高速增長(zhǎng)率���。預(yù)計(jì)到2025年�,市場(chǎng)規(guī)模有望達(dá)到305.39億美元����。雖然中國(guó)市場(chǎng)起步較晚�,但在整體市場(chǎng)發(fā)展的推動(dòng)下,在政策推動(dòng)����、 醫(yī)學(xué)技術(shù)進(jìn)步��、監(jiān)管日益完善等多重因素影響下��,2025年有望激增到 178.85億元��。
圖一|全球以及中國(guó)CGT的市場(chǎng)規(guī)模

在全球及中國(guó)生物醫(yī)藥市場(chǎng)均保持高速增長(zhǎng)的大背景下���,在技術(shù)創(chuàng)新不斷涌現(xiàn)、供應(yīng)鏈優(yōu)勢(shì)不斷凸顯的支撐下���,國(guó)內(nèi)生物醫(yī)藥企業(yè)開(kāi)始在海外布局�����,拓展國(guó)際市場(chǎng)����。特別是在我國(guó)藥監(jiān)部門(mén)加入ICH(國(guó)際人用藥品注冊(cè)技術(shù)協(xié)調(diào)會(huì))后��,為了實(shí)現(xiàn)與國(guó)際接軌��,越來(lái)越多的本土藥企按照 FDA 的標(biāo)準(zhǔn)研發(fā)和申報(bào)新藥�;同時(shí)境外的新藥也加速在中國(guó)落地,這也推動(dòng)了中美雙報(bào)的發(fā)展。
中美雙報(bào)是指用同一套研究資料��,同時(shí)或分階段在中美兩國(guó)分別進(jìn)行申報(bào)����,同時(shí)敲響兩地市場(chǎng)的大門(mén)。但是“中美雙報(bào)”并不是簡(jiǎn)單地把一份資料分別遞送給NMPA和FDA����,而是在ICH框架下同步開(kāi)展在兩國(guó)的新藥開(kāi)發(fā),并在研發(fā)中盡可能同時(shí)滿(mǎn)足兩國(guó)的法規(guī)要求�����。盡管中美申報(bào)法規(guī)基本一致�,但是仍然有很大的差異。因此要制定相對(duì)完善的申報(bào)策略��,需要企業(yè)在申報(bào)過(guò)程中結(jié)合具體的產(chǎn)品特點(diǎn)���、患者種族的差異以及中美法規(guī)監(jiān)督部門(mén)對(duì)申報(bào)資料的要求等情況具體分析�����。本文以中國(guó)的II類(lèi)、美國(guó)的Type B溝通交流會(huì)議書(shū)面回復(fù)和新藥臨床試驗(yàn)申(Investigational New Drug Application ����,IND)為例��,對(duì)兩者的區(qū)別進(jìn)行了簡(jiǎn)單地梳理����。
申報(bào)流程
相同處:原則上�,先與監(jiān)管方進(jìn)行pre-IND溝通交流會(huì)議,監(jiān)管方就溝通交流問(wèn)題做出建議后���,申請(qǐng)人根據(jù)監(jiān)管建議�,補(bǔ)充相關(guān)研究后提交IND申請(qǐng)���。
不同處:
中國(guó):
申請(qǐng)人經(jīng)“申請(qǐng)人之窗”提交《溝通交流會(huì)議申請(qǐng)表》*和《溝通交流會(huì)議資料》**
CDE在收到申請(qǐng)后3日內(nèi)完成初步審核���,符合要求者送達(dá)專(zhuān)業(yè)審評(píng)團(tuán)隊(duì)
II類(lèi)會(huì)議回復(fù)時(shí)限為60個(gè)工作日。經(jīng)“申請(qǐng)人之窗”推送pre-IND問(wèn)題答復(fù)
IND資料受理后60個(gè)工作日�,若臨床試驗(yàn)申請(qǐng)通過(guò),CDE將經(jīng)“申請(qǐng)人之窗”推送“臨床試驗(yàn)批準(zhǔn)通知書(shū)”
圖二|中國(guó)preIND+IND申報(bào)流程

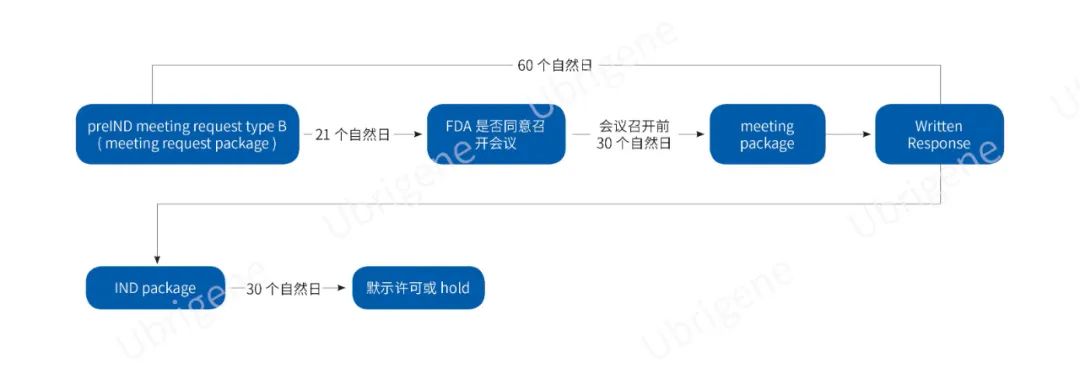
美國(guó):
提前一周申請(qǐng)pre-IND編號(hào)
eCTD交preIND Request(問(wèn)題清單不超過(guò)12個(gè)問(wèn)題)后21天收到FDA回信
第30天提供preIND package(所有問(wèn)題的背景信息���,包括所有附件不超過(guò)150頁(yè))
第60天收到書(shū)面回復(fù)或電話(huà)會(huì)議����,電話(huà)會(huì)議將在結(jié)束1個(gè)月左右收到Meeting Minute
一個(gè)產(chǎn)品或多個(gè)高度相似的產(chǎn)品只有1次pre-IND的機(jī)會(huì),需要進(jìn)一步溝通的問(wèn)題可以要求Type D會(huì)議�,但是Type D只有30%~50%的批準(zhǔn)概率
IND提交30天后,F(xiàn)DA不回信就代表通過(guò)
圖三|FDA pre-IND+IND申報(bào)流程
申報(bào)資料內(nèi)容
相同處:格式和內(nèi)容參考ICH M4 的要求���。
不同處:
M1是區(qū)域行政信息���,中美不太一致。
中國(guó)的M1包括說(shuō)明函���、申請(qǐng)表�、preIND回復(fù)�、臨床藥物說(shuō)明書(shū)等。
美國(guó)的M1包含行政表格(1571�、3674)、US Agent信息���、DMF引用信����、IB���、preIND回復(fù)�、環(huán)境評(píng)估(一般IND直接交豁免信)等����。
中國(guó)臨床試驗(yàn)相關(guān)資料1.3.5主要包括臨床試驗(yàn)方案、知情同意書(shū)����、研究者手冊(cè)。
美國(guó)臨床試驗(yàn)資料則在5.3.5.2體現(xiàn)����,包括臨床方案、ICF��、CV����,批準(zhǔn)后交1572表格
申報(bào)遞交形式
中國(guó):以電子形式(光盤(pán))郵寄申報(bào)資料,格式為PDF��,電子簽章��。藥審中心持續(xù)推進(jìn)對(duì)電子申報(bào)普及�,規(guī)范電子光盤(pán)技術(shù)要求規(guī)范����。
美國(guó):按照ICH M8要求���,采用eCTD電子注冊(cè)文檔的形式�����。M2���、M3S、M3P�����、M3A需要拆分至合適的粒度�、M4毒理報(bào)告要求SEND格式、所有報(bào)告要有STF����。小于等于10G的eCTD可以通過(guò)電子提交網(wǎng)關(guān)ESG提交。
生產(chǎn)批次
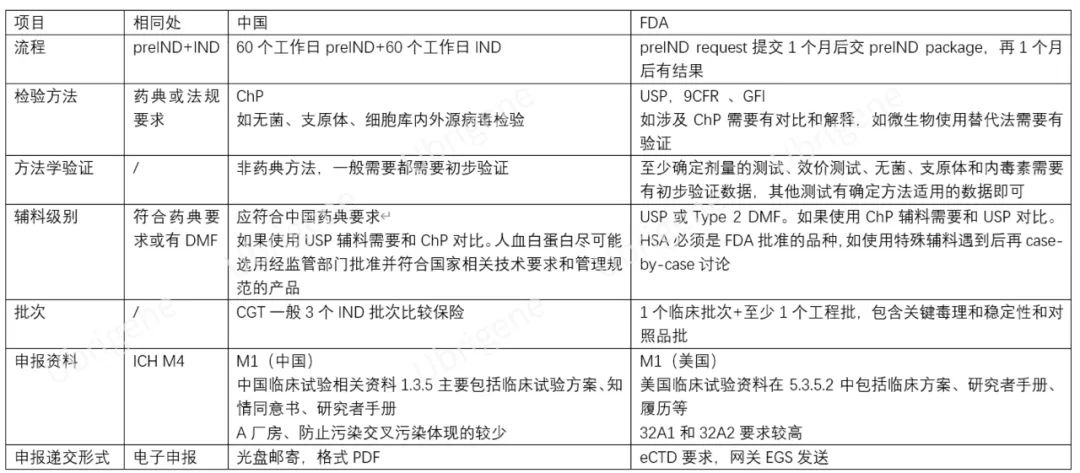
中國(guó):CGT一般3個(gè)IND批次���,對(duì)GMP要求較高�����。
美國(guó):1個(gè)臨床批次+至少1個(gè)工程批��,包含關(guān)鍵毒理和穩(wěn)定性和對(duì)照品批���。I期臨床批次對(duì)GMP要求較低。
生產(chǎn)過(guò)程控制
中國(guó):需要中控試驗(yàn)和中間品穩(wěn)定性方案���,可以不提供數(shù)據(jù)
美國(guó):需要中控和中間品檢測(cè)的數(shù)據(jù)�,當(dāng)有中間品儲(chǔ)存時(shí)要有中間品穩(wěn)定性數(shù)據(jù)�����。
細(xì)胞庫(kù)和病毒庫(kù)
中國(guó):按照中國(guó)藥典三部通則《生物制品生產(chǎn)檢定用動(dòng)物細(xì)胞基質(zhì)制備及質(zhì)量控制》和《生物制品生產(chǎn)檢定用菌毒種管理及質(zhì)量控制》要求執(zhí)行�。
美國(guó):按USP、9 CFR和GFI完成外源因子測(cè)試����。
輔料級(jí)別
中國(guó):應(yīng)符合中國(guó)藥典要求。如果使用USP輔料需要和ChP對(duì)比�����。人血白蛋白盡可能選用經(jīng)監(jiān)管部門(mén)批準(zhǔn)并符合國(guó)家相關(guān)技術(shù)要求和管理規(guī)范的產(chǎn)品。
美國(guó):應(yīng)為USP或Type 2 DMF���。如果使用ChP輔料需要和USP對(duì)比��。HSA必須是FDA批準(zhǔn)的品種����。如果有特殊輔料(比如mRNA用全新結(jié)構(gòu)的脂質(zhì)體)����,需要以DMF或在3.2.A.3中遞交完整資料。
檢驗(yàn)方法
中國(guó):應(yīng)符合中國(guó)藥典要求�����。如無(wú)菌���、支原體��、細(xì)胞內(nèi)外源病毒檢驗(yàn)應(yīng)符合中國(guó)藥典要求�����。
美國(guó):應(yīng)符合美國(guó)藥典要��。如無(wú)菌����、支原體、細(xì)胞內(nèi)外源病毒檢驗(yàn)應(yīng)符合美國(guó)藥典��、9CFR要求����。如涉及ChP需要有對(duì)比和解釋?zhuān)缥⑸锸褂锰娲ㄐ枰序?yàn)證��。
放行標(biāo)準(zhǔn)
相同處:一般包括鑒別����、純度和雜質(zhì)、含量���、生物學(xué)活性�����、安全性和制劑相關(guān)指標(biāo)�����。
美國(guó):異常毒性(general toxicity)只有在極少的情況下需要�。
穩(wěn)定性
中國(guó):運(yùn)輸穩(wěn)定性應(yīng)能覆蓋至國(guó)內(nèi)醫(yī)療機(jī)構(gòu)的運(yùn)輸方式和時(shí)長(zhǎng)。
美國(guó):應(yīng)有運(yùn)輸條件下(運(yùn)輸穩(wěn)定性應(yīng)可以模擬運(yùn)輸至國(guó)外的運(yùn)輸方式和時(shí)長(zhǎng))的穩(wěn)定性數(shù)據(jù)���,CGT產(chǎn)品需要有給藥器械和給藥條件下的穩(wěn)定性數(shù)據(jù)�。
方法學(xué)驗(yàn)證
中國(guó):非藥典方法���,一般需要都需要初步驗(yàn)證��。在藥品開(kāi)發(fā)初期�����,不需要提交全面完整的分析方法驗(yàn)證資料����,但至少應(yīng)提供方法的專(zhuān)屬性����、靈敏度等關(guān)鍵項(xiàng)目的驗(yàn)證信息。
美國(guó):至少確定劑量的測(cè)試����、效價(jià)測(cè)試����、無(wú)菌���、支原體和內(nèi)毒素需要有初步驗(yàn)證數(shù)據(jù)�����,其他測(cè)試有確定方法適用的數(shù)據(jù)即可�����。
包裝容器
中國(guó):優(yōu)先選擇在“原輔包登記平臺(tái)”上登記的包材。
美國(guó):可能的話(huà)提供Type III DMF
小結(jié)
自2017年我國(guó)加入ICH以來(lái)�����,藥品注冊(cè)管理制度加速與國(guó)際接軌��,也推動(dòng)了中美雙報(bào)的發(fā)展���。中美雙報(bào)不僅能幫助本土企業(yè)的產(chǎn)品在更多國(guó)家尤其是發(fā)達(dá)國(guó)家市場(chǎng)上市�,為企業(yè)提供更大的市場(chǎng)和更多的利益,還可以加快產(chǎn)品研發(fā)速度���,培養(yǎng)國(guó)際化臨床研究團(tuán)隊(duì)����,并大幅度地提升本土企業(yè)的國(guó)際影響力���。
然而���,盡管中國(guó)NMPA和美國(guó)FDA在有著大致相同的審核方向,中美雙報(bào)仍然有很大的挑戰(zhàn)性����。
表一|中國(guó)的II類(lèi)、美國(guó)的Type B溝通交流會(huì)議書(shū)面回復(fù)和IND申請(qǐng)比較
(來(lái)源:醫(yī)前沿)