原料藥或制劑的穩(wěn)定性[1]是指其保持物理�����、化學(xué)����、生物學(xué)和微生物學(xué)特性的能力�。通過設(shè)計(jì)試驗(yàn)獲得原料藥或制劑的質(zhì)量特性在各種環(huán)境因素的影響下隨時(shí)間變化的規(guī)律,并據(jù)此為藥品的處方���、工藝��、包裝����、貯藏條件和有效期/復(fù)檢期的確定提供支持性信息����。藥品的穩(wěn)定性是滿足其臨床使用及流通必須具備的特性,也是藥品質(zhì)量研究的重要內(nèi)容�,貫穿于藥品的整個(gè)生命周期。為規(guī)范藥品穩(wěn)定性研究��,國內(nèi)外各監(jiān)管機(jī)構(gòu)發(fā)布了一系列的法律法規(guī)和指南�,以期引導(dǎo)企業(yè)開展良好的穩(wěn)定性研究,確保穩(wěn)定性數(shù)據(jù)能夠真實(shí)反映藥品在儲(chǔ)存期間的質(zhì)量�。同時(shí),國內(nèi)外監(jiān)管機(jī)構(gòu)在開展藥品檢查時(shí)����,也發(fā)現(xiàn)了企業(yè)在開展穩(wěn)定性研究時(shí)存在一些違反法律法規(guī)或指南文件要求的現(xiàn)象。本文旨在通過分析國內(nèi)外相關(guān)法律法規(guī)中對藥品穩(wěn)定性研究的要求���,結(jié)合近年國內(nèi)外監(jiān)管機(jī)構(gòu)/組織在檢查過程中發(fā)現(xiàn)的有關(guān)缺陷�����,為穩(wěn)定性試驗(yàn)的開展和檢查提供參考和借鑒��。
1.穩(wěn)定性研究相關(guān)要求
1.1 技術(shù)要求
原國家食品藥品監(jiān)督管理總局[ 1 ]��、國家藥典委員會(huì)[2]以及人用藥品技術(shù)要求國際協(xié)調(diào)理事會(huì)(The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use�,ICH)發(fā)布的指導(dǎo)原則[3]均列舉了新原料藥及其制劑穩(wěn)定性試驗(yàn)的主要研究內(nèi)容���,為穩(wěn)定性研究的開展提供了指導(dǎo)��,企業(yè)可結(jié)合品種特性����,參考指導(dǎo)原則的相關(guān)要求,設(shè)計(jì)并開展穩(wěn)定性研究���。
1.2 規(guī)范性要求
我國《藥品生產(chǎn)質(zhì)量管理規(guī)范》(2010年修訂)[ 4 ](以下簡稱GMP)�、歐洲藥品管理局(European Medicines Agency�,EMA)GMP[5]、藥品檢查合作計(jì)劃(Pharmaceutical Inspection Cooperation Scheme��,PIC/S)GMP[6]����、美國食品藥品管理局(Food and Drug Administration,F(xiàn)DA)cGMP(Current Good Manufacture Practices)[7]和世界衛(wèi)生組織(World Health Organization��,WHO)GMP[8]等均針對藥品的穩(wěn)定性研究提出了相關(guān)要求���。其中我國�����、EMA和PIC/S的GMP中對穩(wěn)定性考察的描述較為詳細(xì)��,要求基本一致���,F(xiàn)DA和WHO的要求相近�,詳見表1�����。

2011年����,原國家食品藥品監(jiān)督管理局發(fā)布了原料藥附錄作為GMP的配套文件����,ICH Q7活性藥物成分的GMP指南/問答也已于2015年12月31日在我國落地實(shí)施。我國上市的原料藥在開展穩(wěn)定性試驗(yàn)時(shí)�,除滿足上述要求外,還需符合原料藥附錄及ICH Q7[9]等的相關(guān)要求����。
此外,質(zhì)量管理體系[10]通過對產(chǎn)品的整個(gè)生命周期(包括藥品研發(fā)�����、技術(shù)轉(zhuǎn)移��、商業(yè)化生產(chǎn)和產(chǎn)品終止等)中影響產(chǎn)品質(zhì)量的所有因素進(jìn)行管理����,以期為產(chǎn)品的質(zhì)量提供全面有效的保證����。穩(wěn)定性試驗(yàn)的開展除符合上述技術(shù)要求外��,還需以企業(yè)良好的質(zhì)量管理體系為基礎(chǔ)�。
1.3 現(xiàn)場檢查相關(guān)要求
除上述要求外,國內(nèi)外監(jiān)管機(jī)構(gòu)/組織也制定了相應(yīng)的檢查工作程序或合規(guī)手冊�,明確了穩(wěn)定性的檢查范圍、要求及風(fēng)險(xiǎn)的判定原則�。見表2。

根據(jù)《藥品注冊核查要點(diǎn)與判定原則(藥學(xué)研制和生產(chǎn)現(xiàn)場)》(試行)[11]�����,穩(wěn)定性研究及承擔(dān)該研究內(nèi)容的機(jī)構(gòu)均是藥品注冊核查的重點(diǎn)�。2022年,美國FDA發(fā)布的cGMP 7346.832 批準(zhǔn)前檢查[12]中也提到了穩(wěn)定性的要求和核查重點(diǎn)����,與我國的《藥品注冊核查要點(diǎn)與判定原則(藥學(xué)研制和生產(chǎn)現(xiàn)場)》(試行)基本一致。該合規(guī)手冊還提出了在檢查時(shí)應(yīng)關(guān)注異常事件的處理����,這些問題通常反映了企業(yè)潛在的數(shù)據(jù)可靠性風(fēng)險(xiǎn)����。
2.檢查中常見的穩(wěn)定性相關(guān)缺陷
《藥品生產(chǎn)現(xiàn)場檢查風(fēng)險(xiǎn)評定指導(dǎo)原則》[13]中列舉了穩(wěn)定性考察相關(guān)的缺陷��,并根據(jù)其缺陷嚴(yán)重程度以及產(chǎn)品風(fēng)險(xiǎn)分類��,對其風(fēng)險(xiǎn)級別進(jìn)行了判定��。FDA發(fā)布的cGMP 7346.832 批準(zhǔn)前檢查[12]和cGMP 7356.002 藥品生產(chǎn)檢查[14]�,也列舉了可能存在的穩(wěn)定性相關(guān)的嚴(yán)重缺陷�,檢查組可基于發(fā)現(xiàn)問題的程度,將缺陷項(xiàng)列入483表格����,或據(jù)此引發(fā)官方行動(dòng)指示(Official Action Indicated,OAI)��,詳見表3�����。

2.1 我國組織實(shí)施的檢查
2.1.1 注冊核查
通過對國家藥品監(jiān)督管理局食品藥品審核查驗(yàn)中心(以下簡稱核查中心)2020至2022年開展的境內(nèi)企業(yè)化學(xué)藥品注冊核查有關(guān)缺陷進(jìn)行梳理����、統(tǒng)計(jì)和分析���,穩(wěn)定性研究相關(guān)缺陷約占發(fā)現(xiàn)缺陷總條款數(shù)的6%左右,其中針對研制單位/部門提出的約占36%���。涉及條款主要集中在GMP正文第233條��、第234條����、第238條�����。此外�,占比較高的還包括正文第223條和第250條,主要體現(xiàn)在穩(wěn)定性試驗(yàn)規(guī)程/方案的制定與執(zhí)行�、數(shù)據(jù)可靠性(實(shí)驗(yàn)室異常事件、穩(wěn)定性試驗(yàn)數(shù)據(jù)的獲得或處理)��、穩(wěn)定性試驗(yàn)設(shè)備或設(shè)施�、檢驗(yàn)記錄等方面的不足。我國藥品注冊核查中穩(wěn)定性相關(guān)缺陷占比情況見圖1��。
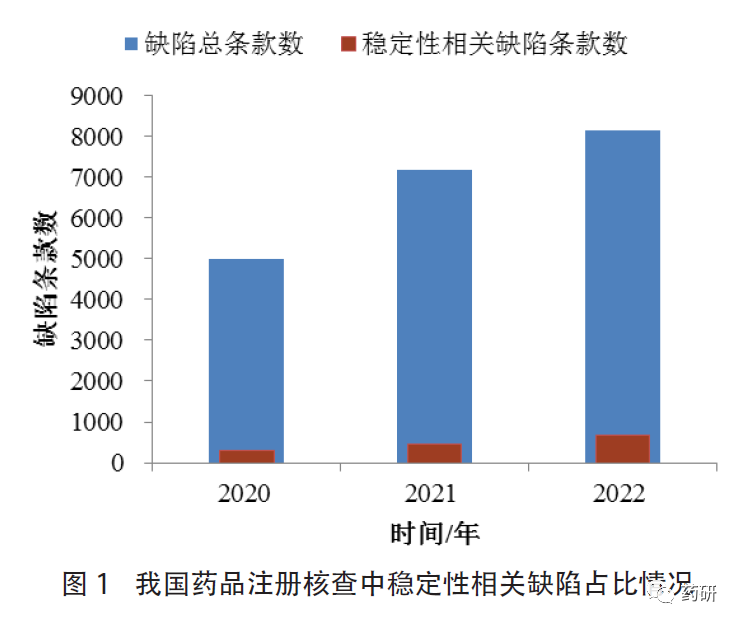
(1)穩(wěn)定性試驗(yàn)規(guī)程/方案的制定與執(zhí)行:GMP(2010年版)正文第234條對穩(wěn)定性試驗(yàn)方案提出了較強(qiáng)的實(shí)操性要求,依據(jù)該條款提出的缺陷較多��。例如未注明穩(wěn)定性試驗(yàn)考察項(xiàng)目的可接受標(biāo)準(zhǔn)�、檢驗(yàn)方法依據(jù)或顯著差異的判定標(biāo)準(zhǔn),缺少部分關(guān)鍵檢驗(yàn)項(xiàng)目但未給出充分理由�。隨著注冊申報(bào)品種的增多,工作量的增加及可能存在的不可抗力等因素的影響����,部分企業(yè)還存在樣品生產(chǎn)完成后未能及時(shí)放樣,或未按規(guī)定的時(shí)間點(diǎn)及時(shí)取樣�、檢驗(yàn)。此外����,藥品注冊申報(bào)期間��,質(zhì)量標(biāo)準(zhǔn)可能會(huì)根據(jù)審評的要求進(jìn)行修訂����,企業(yè)應(yīng)根據(jù)質(zhì)量標(biāo)準(zhǔn)的變更情況評估是否需要修訂穩(wěn)定性試驗(yàn)方案。
(2)數(shù)據(jù)可靠性:主要體現(xiàn)在數(shù)據(jù)獲得或處理方式不合理(例如未按規(guī)定的標(biāo)準(zhǔn)或方法檢驗(yàn)��、計(jì)算或積分不合理)����、異常事件的處理不當(dāng)(例如無故復(fù)測���、未開展調(diào)查��、調(diào)查不充分或未采取有效的糾正與預(yù)防措施)����、數(shù)據(jù)管理存在不足(例如未按規(guī)定有效備份���、未開啟審計(jì)追蹤�、權(quán)限設(shè)置不合理)等方面����。
(3)穩(wěn)定性試驗(yàn)設(shè)備或設(shè)施:穩(wěn)定性試驗(yàn)設(shè)備或設(shè)施是確保穩(wěn)定性研究環(huán)境符合要求的關(guān)鍵,常見問題集中在持續(xù)穩(wěn)定提供符合要求的考察環(huán)境的能力上��。ICH Q1A (R2)[3]指出企業(yè)應(yīng)記錄設(shè)備失控而導(dǎo)致的偏離的影響���,如已證明影響穩(wěn)定性結(jié)果���,則需報(bào)告。以某次現(xiàn)場檢查為例�����,承擔(dān)穩(wěn)定性研究的研制單位未對恒溫恒濕穩(wěn)定性試驗(yàn)箱進(jìn)行溫濕度分布驗(yàn)證或校準(zhǔn);每日僅打印記錄2個(gè)時(shí)間點(diǎn)的溫濕度���,以打印的溫濕度數(shù)據(jù)作為原始數(shù)據(jù)����,但未對已更換的溫濕度打印記錄條進(jìn)行保存����;穩(wěn)定性試驗(yàn)箱連續(xù)多日出現(xiàn)溫濕度異常,但企業(yè)的《溫濕度巡檢記錄》顯示溫濕度均為正常�����;未明確溫濕度異常情況的處理措施�?���?傮w上,在人員�����、設(shè)備和管理等多方面不受控。
(4)檢驗(yàn)記錄:常見的缺陷包括缺少關(guān)鍵原始記錄�、記錄不及時(shí)、樣品數(shù)量與實(shí)際不符等����。此外,常見的缺陷還涉及人員職責(zé)�����、變更控制����、包裝、文件管理以及委托研究等���。例如未明確穩(wěn)定性試驗(yàn)相關(guān)人員職責(zé)(尤其是質(zhì)量受權(quán)人)或履職不充分���;未將穩(wěn)定性試驗(yàn)納入變更管理,重大變更未列入穩(wěn)定性考察��;試驗(yàn)樣品未采用市售包裝�,再包裝后藥品缺少有效期確定的依據(jù);委托研究未簽訂書面協(xié)議����、申請人/持有人履職不充分��、缺少對受托方的審計(jì)���、未對委托方和受托方檢測結(jié)果的差異開展分析或調(diào)查等。
2.1.2 上市后檢查
在對已上市化學(xué)藥品開展的跟蹤檢查和飛行檢查中��,還存在未按規(guī)定開展持續(xù)穩(wěn)定性考察�、年度回顧時(shí)未對穩(wěn)定性考察的結(jié)果及不良趨勢進(jìn)行分析調(diào)查或處理,甚至將穩(wěn)定性考察超標(biāo)數(shù)據(jù)記錄為合格的嚴(yán)重違反GMP的情形��。
2.1.3 境外檢查
核查中心近年組織的境外檢查中�����,約三分之一的被檢查單位在穩(wěn)定性研究方面存在不足��。例如未針對中國市場的產(chǎn)品開展持續(xù)穩(wěn)定性考察�����、質(zhì)量受權(quán)人及質(zhì)量部門負(fù)責(zé)人的崗位職責(zé)中均未包括穩(wěn)定性考察相關(guān)職責(zé)�、待包裝產(chǎn)品穩(wěn)定性數(shù)據(jù)不足等�。
2.2 FDA 檢查中常見的穩(wěn)定性相關(guān)缺陷
檢索FDA 2020財(cái)年至2022財(cái)年簽發(fā)的藥品相關(guān)483表格[15]�,累計(jì)分別為349���、215�����、466份�,與21CFR 211.137有效期或21 CFR 211.166穩(wěn)定性試驗(yàn)相關(guān)的分別有47條�����、42條和91條�����,詳見圖2���。

以“stability”為檢索詞檢索FDA 2020至2022年發(fā)出的警告信[16]�,涉及條款除21 CFR 211.137有效期和21 CFR 211.166穩(wěn)定性試驗(yàn)外����,還包括21CFR 211.192生產(chǎn)記錄審核(異常事件的調(diào)查)、21 CFR 211.22質(zhì)量管理部門職責(zé)(受托單位的監(jiān)管����、變更和人員職責(zé))和21 CFR 211.68自動(dòng)化��、機(jī)械化及電子設(shè)備(穩(wěn)定性試驗(yàn)設(shè)備或設(shè)施)等�,詳見圖3�����。
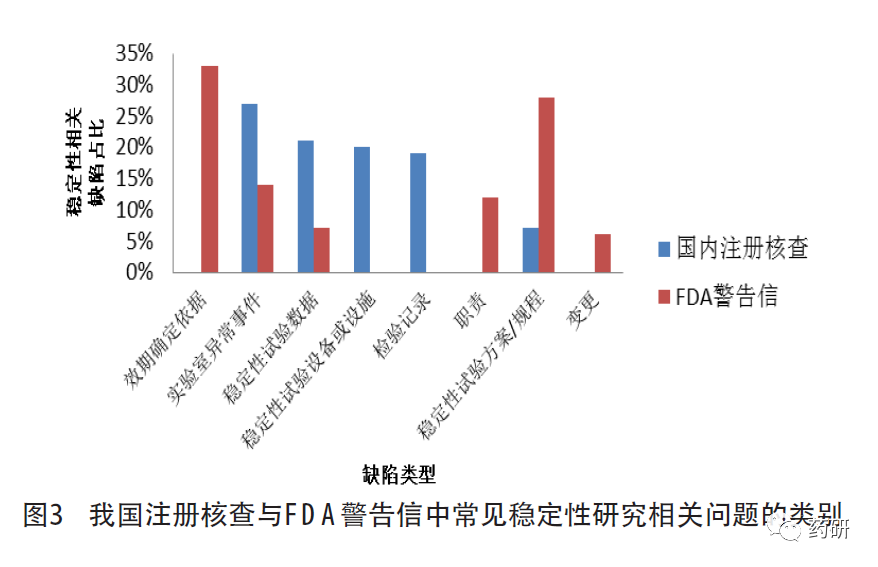
對比我國注冊核查與FDA警告信中發(fā)現(xiàn)的穩(wěn)定性試驗(yàn)的相關(guān)缺陷����,穩(wěn)定性試驗(yàn)規(guī)程/方案的制定與執(zhí)行不到位、數(shù)據(jù)可靠性(實(shí)驗(yàn)室異常事件�����、穩(wěn)定性數(shù)據(jù)的獲得或處理)管理不足均占比較高���。我國注冊核查中����,因委托研制單位開展穩(wěn)定性研究的占比較高�����,穩(wěn)定性試驗(yàn)設(shè)備或設(shè)施和檢驗(yàn)記錄方面的缺陷占比較FDA警告信高����。與我國注冊核查不同,F(xiàn)DA警告信中有效期的確定依據(jù)不足����、職責(zé)未明確方面的缺陷占比較高,這可能與新冠肺炎疫情期間手消毒產(chǎn)品以及美國國內(nèi)生產(chǎn)的非處方藥產(chǎn)品的警告信增加有關(guān)����。此外,F(xiàn)DA警告信中變更相關(guān)的缺陷比例也相對較高�,主要與上市后變更相關(guān),即變更后未按要求開展穩(wěn)定性研究���,這與我國已上市化學(xué)藥品開展的跟蹤檢查和飛行檢查中發(fā)現(xiàn)的情況基本一致�。
2.3 國外監(jiān)管機(jī)構(gòu)/組織對我國企業(yè)開展的檢查
近年來國外藥品監(jiān)管機(jī)構(gòu)包括WHO����、FDA、歐洲藥品質(zhì)量管理局(European Directorate for the Quality of Medicines & HealthCare)[17]對我國境內(nèi)藥品生產(chǎn)企業(yè)開展的檢查中����,穩(wěn)定性相關(guān)缺陷占比約為6.9%�,主要集中在異常事件的處理�、穩(wěn)定性試驗(yàn)規(guī)程/方案、數(shù)據(jù)可靠性和穩(wěn)定性試驗(yàn)設(shè)備或設(shè)施等方面��,與我國藥品注冊核查發(fā)現(xiàn)的情況基本一致�。此外,同樣發(fā)現(xiàn)部分企業(yè)在變更管理方面存在不足����,即未對工藝驗(yàn)證批次、或部分偏差及返工批次開展穩(wěn)定性試驗(yàn)�����。
3.案例分析和建議
結(jié)合國內(nèi)外監(jiān)管機(jī)構(gòu)/組織現(xiàn)場檢查常見的穩(wěn)定性研究相關(guān)缺陷��,以及我國委托研制單位/部門承擔(dān)穩(wěn)定性研究的占比逐漸升高的現(xiàn)狀����,本文將通過案例近一步分析和討論穩(wěn)定性研究中需重點(diǎn)關(guān)注的內(nèi)容。
3.1 案例分析
3.1.1 穩(wěn)定性試驗(yàn)規(guī)程/方案的制定與執(zhí)行
穩(wěn)定性試驗(yàn)規(guī)程/方案是開展穩(wěn)定性研究的基礎(chǔ)和依據(jù)���,而良好的執(zhí)行是獲得真實(shí)����、可靠數(shù)據(jù)的有力保障。穩(wěn)定性研究應(yīng)考察在貯藏期間易發(fā)生變化����、可能影響其質(zhì)量�、安全性和有效性的項(xiàng)目,考察時(shí)間及考察頻率也直接影響穩(wěn)定性結(jié)果的統(tǒng)計(jì)與判定��。以核查發(fā)現(xiàn)缺陷為例:某片劑的含量限度為95.0%~105.0%����,放行檢測含量為103.5%,4個(gè)月后放樣進(jìn)行穩(wěn)定性研究�����,重新檢測的含量為98.2%�����,超出了申請人確定的顯著差異的判定標(biāo)準(zhǔn)�。企業(yè)在穩(wěn)定性試驗(yàn)規(guī)程/方案中,未明確穩(wěn)定性0天的放樣時(shí)間�����、樣品的暫存條件和0天數(shù)據(jù)的確定依據(jù),在重新檢測后���,也未對2次檢測結(jié)果的差異進(jìn)行調(diào)查分析��,直接以第2次的含量檢測結(jié)果作為穩(wěn)定性0天的數(shù)據(jù)����。ICH Q1A(R2)[2]提到“考察時(shí)間�,尤其是在注冊階段,可能會(huì)影響數(shù)據(jù)的統(tǒng)計(jì)分析��,趨勢判斷���,甚至有效期的確定”���。在申報(bào)過程中,受批次和考察時(shí)間的限制�,穩(wěn)定性數(shù)據(jù)較少,此情況可能掩蓋了加速穩(wěn)定性考察期間含量的變化趨勢��,不利于有效期的外推或判定�����。
企業(yè)在制定穩(wěn)定性相關(guān)文件時(shí),可根據(jù)產(chǎn)品的特性��,對穩(wěn)定性考察時(shí)間點(diǎn)(包括起點(diǎn)�、試驗(yàn)間隔時(shí)間、終點(diǎn))進(jìn)行規(guī)定�,評估并確定可接受的范圍,通過風(fēng)險(xiǎn)管理對可能出現(xiàn)的異常情況作出預(yù)案���。
3.1.2 數(shù)據(jù)可靠性
《藥品注冊核查要點(diǎn)與判定原則(藥學(xué)研制和生產(chǎn)現(xiàn)場)》(試行)指出,穩(wěn)定性研究所涉及的數(shù)據(jù)應(yīng)當(dāng)能溯源����,并完整可靠,方法學(xué)驗(yàn)證及之后影響產(chǎn)品質(zhì)量和穩(wěn)定性數(shù)據(jù)評價(jià)的研究數(shù)據(jù)尤為重要���。該原則中����,多項(xiàng)判定為“不通過”的情形��,均與數(shù)據(jù)可靠性有關(guān)?���,F(xiàn)場檢查發(fā)現(xiàn)�,長期穩(wěn)定性試驗(yàn)有關(guān)物質(zhì)檢測時(shí)���,有企業(yè)直接通過設(shè)置禁止積分時(shí)間段��,將超過質(zhì)量標(biāo)準(zhǔn)規(guī)定的最大未知單雜禁止積分而不報(bào)告該雜質(zhì)�,或通過調(diào)整積分參數(shù)(例如斜率�����、峰的起止時(shí)間)等造成雜質(zhì)檢測數(shù)據(jù)偏低���。這些均提示企業(yè)可能存在嚴(yán)重的數(shù)據(jù)可靠性問題����,并反映了企業(yè)在質(zhì)量管理方面的不足��。此外��,穩(wěn)定性考察期間出現(xiàn)的異常數(shù)據(jù)�,可能是對樣品當(dāng)下質(zhì)量或趨勢的反映。企業(yè)應(yīng)根據(jù)我國GMP和ICH等指導(dǎo)原則的要求��,對超標(biāo)和異常趨勢進(jìn)行調(diào)查,并考慮可能對已上市產(chǎn)品造成的影響�����,必要時(shí)應(yīng)當(dāng)實(shí)施召回�����,并報(bào)告當(dāng)?shù)厮幤繁O(jiān)督管理部門����。在對已上市產(chǎn)品開展的飛行檢查中發(fā)現(xiàn),某產(chǎn)品有關(guān)物質(zhì)(最大單雜限度為1.5%)在穩(wěn)定性考察期間呈上升趨勢����,其中1個(gè)批次的產(chǎn)品最大單雜長期6月(1.3%)和長期9月(1.4%)檢測結(jié)果已經(jīng)超出原有考察批次的趨勢�����,且已接近上限�����。企業(yè)未根據(jù)歷史批次的考察數(shù)據(jù)對該情況進(jìn)行對比分析�����,仍僅以是否符合質(zhì)量標(biāo)準(zhǔn)要求為判定依據(jù),掩蓋了已上市產(chǎn)品的質(zhì)量風(fēng)險(xiǎn)���,不利于及時(shí)采取措施���。最終長期12月最大單雜檢測結(jié)果達(dá)到限度(1.5%),長期18月檢測結(jié)果超出標(biāo)準(zhǔn)規(guī)定的限度����,現(xiàn)場檢查提示該企業(yè)質(zhì)量管理系統(tǒng)不能有效運(yùn)行,不符合GMP的要求���。
針對貯藏期間易發(fā)生變化的項(xiàng)目��,企業(yè)可根據(jù)產(chǎn)品特性�����,采取設(shè)置警戒限或行動(dòng)限��,縮短試驗(yàn)間隔�����,增加數(shù)據(jù)對比方式(例如批內(nèi)和批間)等措施����,必要時(shí)重新對工藝進(jìn)行研究,以便及時(shí)發(fā)現(xiàn)并捕捉穩(wěn)定性試驗(yàn)過程中的趨勢和異常����,確保上市產(chǎn)品符合質(zhì)量標(biāo)準(zhǔn)的要求。
3.1.3 委托研究
研制單位/部門的出現(xiàn)和介入為藥品注冊申報(bào)提供了很好的技術(shù)平臺��,提高了研發(fā)效率�����,但也帶來了新的挑戰(zhàn)�,尤其是合規(guī)性方面。2010年修訂的GMP規(guī)定����,關(guān)鍵人員���,尤其是質(zhì)量受權(quán)人���,應(yīng)當(dāng)了解持續(xù)穩(wěn)定性考察的結(jié)果�����。當(dāng)持續(xù)穩(wěn)定性考察不在待包裝產(chǎn)品和成品的生產(chǎn)企業(yè)進(jìn)行時(shí)���,則相關(guān)各方之間應(yīng)當(dāng)有書面協(xié)議。部分申請人在委托進(jìn)行穩(wěn)定性試驗(yàn)時(shí)��,并未簽訂書面協(xié)議�,或未根據(jù)協(xié)議對方案進(jìn)行審批或確認(rèn)。例如現(xiàn)場檢查發(fā)現(xiàn)�,委托方和受托方簽訂的穩(wěn)定性研究委托協(xié)議中包括粒度檢查項(xiàng),但受托方的穩(wěn)定性試驗(yàn)方案中未包括該檢項(xiàng)����,且委托方未按協(xié)議對方案進(jìn)行審核,以至于未發(fā)現(xiàn)該漏檢項(xiàng)目���。再如����,受托單位在對某注射液(三層共擠輸液袋裝)進(jìn)行穩(wěn)定性研究時(shí)����,工藝驗(yàn)證批0天滲透壓為297 mOsmol·kg-1(限度270~320mOsmol·kg-1)��,長期6月失水率為1.39%����,滲透壓為306 mOsmol·kg-1�,長期9月失水率為2.69%,滲透壓為312 mOsmol·kg-1�����,長期12月失水率為3.20%�����,滲透壓為319mOsmol·kg-1���,呈增長趨勢����,且接近標(biāo)準(zhǔn)規(guī)定的上限����,受托方未按照協(xié)議規(guī)定將穩(wěn)定性研究的相關(guān)趨勢告知申請人。現(xiàn)場檢查期間申請人復(fù)測該批次滲透壓為304 mOsmol·kg-1���,隨即采用復(fù)測數(shù)據(jù)作為穩(wěn)定性研究數(shù)據(jù)�。但現(xiàn)有研究數(shù)據(jù)表明失水率��、含量和滲透壓隨考察時(shí)間均呈增長趨勢�����,申請人和受托單位均未結(jié)合品種特性����,及已有的數(shù)據(jù)進(jìn)行趨勢調(diào)查和分析,且申請人在委托研究期間也未落實(shí)主體責(zé)任�,根據(jù)協(xié)議對受托研究單位的資質(zhì)、能力等情況進(jìn)行確認(rèn)及監(jiān)督�。
我國GMP和《藥品注冊核查要點(diǎn)與判定原則(藥學(xué)研制和生產(chǎn)現(xiàn)場)》(試行)均對委托研究提出了相關(guān)要求。企業(yè)可從實(shí)驗(yàn)室內(nèi)部�����、分析方法(例如方法學(xué)驗(yàn)證�、方法學(xué)轉(zhuǎn)移)、樣品及其工藝等方面綜合分析����,查找原因����,并制定管理程序��,規(guī)范此類問題的判定原則和處理方法��。此外��,委托研究時(shí)還需關(guān)注穩(wěn)定性考察樣品的運(yùn)輸和接收����,委托方和受托方質(zhì)量管理文件的兼容性,例如穩(wěn)定性規(guī)程��、變更管理規(guī)程���、實(shí)驗(yàn)室異常事件管理規(guī)程等�,尤其當(dāng)出現(xiàn)不符合質(zhì)量標(biāo)準(zhǔn)的結(jié)果或重大不良趨勢甚至需開展非實(shí)驗(yàn)室因素調(diào)查時(shí)�����,雙方應(yīng)及時(shí)溝通和協(xié)調(diào)�����,明確各方職責(zé),確保委托研究機(jī)構(gòu)產(chǎn)生的數(shù)據(jù)的可靠性�,充分落實(shí)申請人的主體責(zé)任��。
3.2 建議
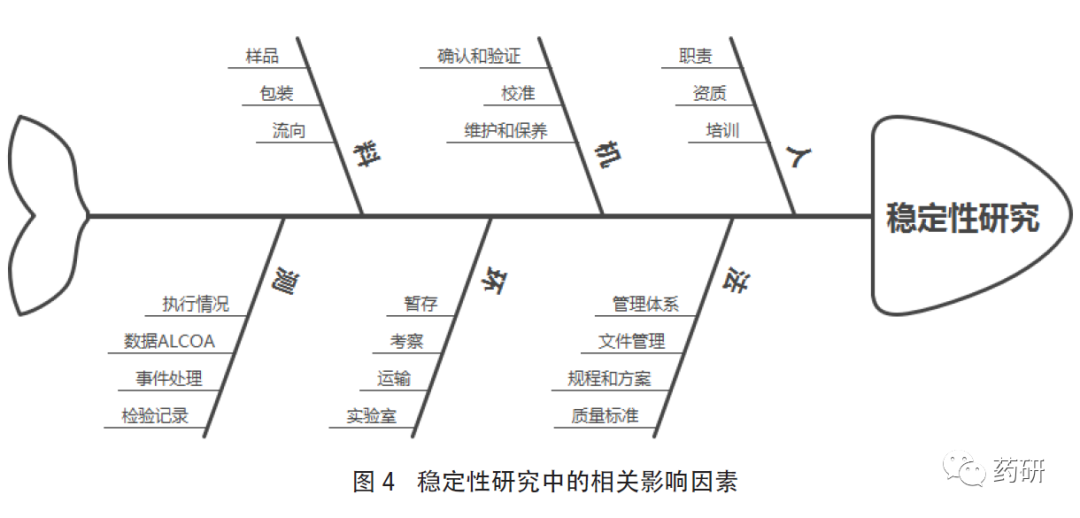
企業(yè)應(yīng)建立標(biāo)準(zhǔn)操作規(guī)程去描述穩(wěn)定性試驗(yàn)的程序和要求�����,并嚴(yán)格執(zhí)行���,任何偏離發(fā)生應(yīng)啟動(dòng)偏差系統(tǒng)進(jìn)行調(diào)查��。企業(yè)在開展穩(wěn)定性試驗(yàn)時(shí)�,可參考國內(nèi)外相關(guān)指導(dǎo)原則�、GMP和《藥品注冊核查要點(diǎn)與判定原則(藥學(xué)研制和生產(chǎn)現(xiàn)場)》(試行)等要求,充分評估自身的組織架構(gòu)和質(zhì)量管理體系����,從人、機(jī)�����、料、法�����、環(huán)��、測等多方面綜合考慮��,做好研究前的準(zhǔn)備工作�����,對可能出現(xiàn)的異常情況進(jìn)行預(yù)判并制定合理的處理措施��。同時(shí)���,根據(jù)產(chǎn)品的目標(biāo)市場��,結(jié)合品種特性����,通過質(zhì)量風(fēng)險(xiǎn)管理評估制定穩(wěn)定
4.結(jié)語
藥品的穩(wěn)定性是滿足其流通及臨床使用必須具備的特性�����,也是藥品質(zhì)量研究的重要內(nèi)容,貫穿于藥品的整個(gè)生命周期�����,對原料藥���、中間產(chǎn)品及制劑的開發(fā)與上市具有重大影響,是制定產(chǎn)品有效期的重要依據(jù)����。本文對比研究了國內(nèi)外相關(guān)法律法規(guī)和指南文件對藥品穩(wěn)定性研究的要求,并對近年來國內(nèi)外監(jiān)管機(jī)構(gòu)/組織在各類檢查中發(fā)現(xiàn)的相關(guān)缺陷進(jìn)行梳理�����、分析和討論�����。企業(yè)在開展穩(wěn)定性研究時(shí)����,可結(jié)合自身質(zhì)量管理體系情況,對比指南文件的要求和本文中提到的常見缺陷��,不斷提升改進(jìn)穩(wěn)定性研究相關(guān)工作的質(zhì)量。同時(shí)�,也希望為藥品檢查工作提供參考和借鑒。
(來源:藥研)