藥品上市后變更管理是藥品全生命周期管理的重要環(huán)節(jié)����。為強(qiáng)化藥品上市后變更管理,強(qiáng)化藥品上市許可持有人(MAH)藥品上市后變更管理主體責(zé)任����,1月13日,國家藥品監(jiān)督管理局正式發(fā)布《藥品上市后變更管理辦法(試行)》(以下簡稱《辦法》)�����。該文件一方面作為藥品監(jiān)管部門藥品注冊(cè)和生產(chǎn)監(jiān)督管理工作的有效銜接�,加強(qiáng)藥品監(jiān)管部門對(duì)藥品上市后監(jiān)管的規(guī)范化管理;另一方面�,逐步完善了新藥品監(jiān)管法規(guī)制度體系建設(shè),在現(xiàn)行藥品注冊(cè)管理法規(guī)體系中具有非常重要的地位���。
本文結(jié)合《辦法》及有關(guān)文件要求���,對(duì)新舊藥品注冊(cè)管理法規(guī)體系中藥品上市后注冊(cè)管理事項(xiàng)變更進(jìn)行橫向?qū)Ρ?,以供參考學(xué)習(xí)�����。
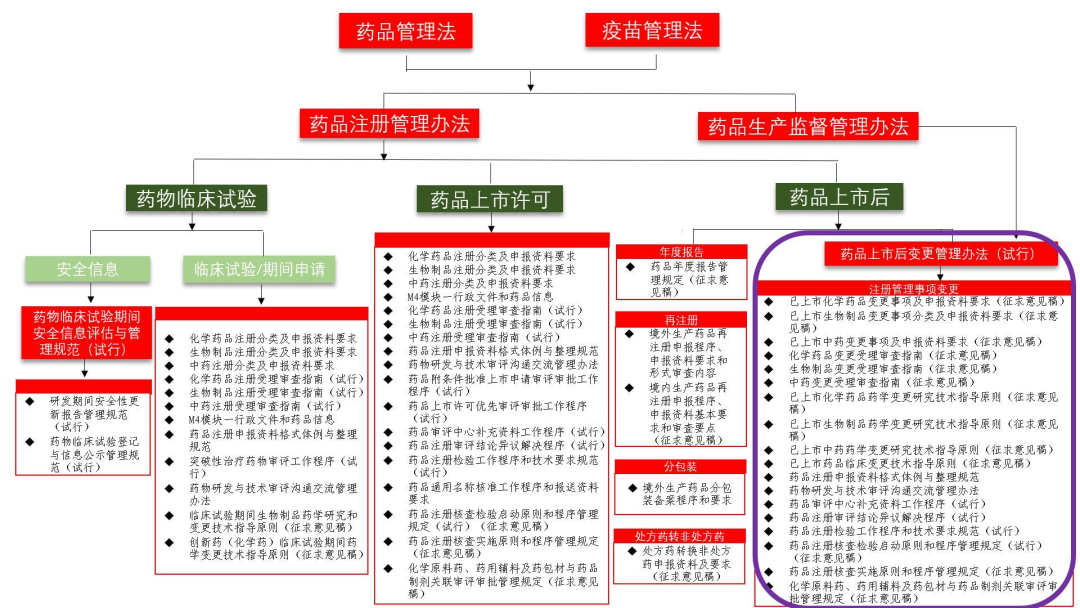
現(xiàn)行藥品注冊(cè)管理主要法規(guī)體系圖
注冊(cè)變更事項(xiàng)分類
原法規(guī)環(huán)境
原《藥品注冊(cè)管理辦法》第八章“補(bǔ)充申請(qǐng)的申報(bào)與審批”第一百一十條規(guī)定:變更研制新藥��、生產(chǎn)藥品和進(jìn)口藥品已獲批準(zhǔn)證明文件及其附件中載明事項(xiàng)的����,應(yīng)當(dāng)提出補(bǔ)充申請(qǐng),并在附件4“藥品補(bǔ)充申請(qǐng)注冊(cè)事項(xiàng)及申報(bào)資料要求”中將補(bǔ)充申請(qǐng)注冊(cè)事項(xiàng)分為��,“國家食品藥品監(jiān)督管理局審批的補(bǔ)充申請(qǐng)事項(xiàng)”“省級(jí)食品藥品監(jiān)督管理部門批準(zhǔn)國家食品藥品監(jiān)督管理局備案或國家食品藥品監(jiān)督管理局直接備案的進(jìn)口藥品補(bǔ)充申請(qǐng)事項(xiàng)”和“省級(jí)食品藥品監(jiān)督管理部門備案的補(bǔ)充申請(qǐng)事項(xiàng)”����。
新法規(guī)環(huán)境
2019年頒布實(shí)施的新修訂《藥品管理法》新增第七章“藥品上市后管理”,其中第七十九條明確�����,對(duì)藥品生產(chǎn)過程中的變更���,按照其對(duì)藥品安全性���、有效性和質(zhì)量可控性的風(fēng)險(xiǎn)和產(chǎn)生影響的程度,實(shí)行分類管理��。新修訂《藥品注冊(cè)管理辦法》第七十七條規(guī)定�����,藥品上市后的變更�����,按照其對(duì)藥品安全性�、有效性和質(zhì)量可控性的風(fēng)險(xiǎn)和產(chǎn)生影響的程度,實(shí)行分類管理��,分為審批類變更�、備案類變更和報(bào)告類變更;第七十八條至第八十條就審批類變更�、備案類變更和報(bào)告類變更情形作出明確規(guī)定。
申報(bào)資料要求
原法規(guī)環(huán)境
原《藥品注冊(cè)管理辦法》附件4中明確了藥品補(bǔ)充申請(qǐng)注冊(cè)事項(xiàng)及申報(bào)資料要求�����,且國家藥監(jiān)局藥審中心(以下簡稱藥審中心)先后發(fā)布《已上市化學(xué)藥品變更研究的技術(shù)指導(dǎo)原則(一)》(2008年)����、《生物制品生產(chǎn)工藝過程變更管理技術(shù)指導(dǎo)原則》(2008年)���、《已上市中藥變更研究技術(shù)指導(dǎo)原則(一)》(2011年)、《疫苗生產(chǎn)場(chǎng)地變更質(zhì)量可比性研究技術(shù)指導(dǎo)原則》(2014年)��、《已上市化學(xué)藥品生產(chǎn)工藝變更研究技術(shù)指導(dǎo)原則》(2017年)�����、《已上市中藥生產(chǎn)工藝變更研究技術(shù)指導(dǎo)原則》(2017年)����,以及《生物制品上市后變更研究技術(shù)指導(dǎo)原則(征求意見稿)》(2017年)和《疫苗上市后生產(chǎn)工藝變更研究技術(shù)指導(dǎo)原則(征求意見稿)》(2019年)。
根據(jù)各補(bǔ)充申請(qǐng)事項(xiàng)的工作流程不同�,審批和備案事項(xiàng)需分別提出申請(qǐng)。
新法規(guī)環(huán)境
新修訂《藥品注冊(cè)管理辦法》發(fā)布后���,藥品監(jiān)管部門在前期已發(fā)布的指導(dǎo)原則基礎(chǔ)上����,參考FDA�、EMA、ICH對(duì)變更的分類以及相關(guān)指導(dǎo)原則�,結(jié)合國內(nèi)研發(fā)與生產(chǎn)現(xiàn)狀,撰寫發(fā)布《已上市化學(xué)藥品變更事項(xiàng)及申報(bào)資料要求(征求意見稿)》《已上市生物制品變更事項(xiàng)分類及申報(bào)資料要求(征求意見稿)》《已上市中藥變更事項(xiàng)及申報(bào)資料要求(征求意見稿)》《化學(xué)藥品變更受理審查指南(征求意見稿)》《生物制品變更受理審查指南(征求意見稿)》《中藥變更受理審查指南(征求意見稿)》《已上市化學(xué)藥品藥學(xué)變更研究技術(shù)指導(dǎo)原則(征求意見稿)》《已上市生物制品藥學(xué)變更研究技術(shù)指導(dǎo)原則(征求意見稿)》《已上市中藥藥學(xué)變更研究技術(shù)指導(dǎo)原則(征求意見稿)》《已上市藥品臨床變更技術(shù)指導(dǎo)原則(征求意見稿)》�,明確各類藥品注冊(cè)變更分類、申報(bào)資料要求和相應(yīng)技術(shù)要求等�����。
《辦法》的政策解讀中明確�,藥品同時(shí)發(fā)生審批類和備案類關(guān)聯(lián)的變更,或備案類變更是以審批類變更獲得批準(zhǔn)為前提時(shí)�,持有人可以將審批類變更和備案類變更合并申報(bào)藥審中心進(jìn)行技術(shù)審評(píng),備案類變更需按照藥品補(bǔ)充申請(qǐng)收費(fèi)標(biāo)準(zhǔn)繳費(fèi)��。
此外�����,《已上市藥品臨床變更技術(shù)指導(dǎo)原則(征求意見稿)》中明確:多種情形臨床變更同時(shí)申報(bào)����,應(yīng)按其中最高類型變更的程序要求進(jìn)行申請(qǐng)和審評(píng)審批;《已上市化學(xué)藥品藥學(xué)變更研究技術(shù)指導(dǎo)原則(征求意見稿)》《已上市生物制品藥學(xué)變更研究技術(shù)指導(dǎo)原則(征求意見稿)》《已上市中藥藥學(xué)變更研究技術(shù)指導(dǎo)原則(征求意見稿)》中均提出“關(guān)聯(lián)變更”總體上需按照技術(shù)要求較高的變更類別進(jìn)行研究�。
關(guān)于持有人變更
原法規(guī)環(huán)境
原《藥品注冊(cè)管理辦法》附件4中明確:“新藥的技術(shù)轉(zhuǎn)讓”屬報(bào)“國家食品藥品監(jiān)督管理局審批的補(bǔ)充申請(qǐng)事項(xiàng)”。依據(jù)《食品藥品監(jiān)管局關(guān)于印發(fā)〈藥品技術(shù)轉(zhuǎn)讓注冊(cè)管理規(guī)定〉的通知》���,申請(qǐng)藥品技術(shù)轉(zhuǎn)讓��,應(yīng)當(dāng)填寫《藥品補(bǔ)充申請(qǐng)表》���,按照補(bǔ)充申請(qǐng)的程序和規(guī)定提交申請(qǐng)���。符合規(guī)定的,發(fā)給《藥品補(bǔ)充申請(qǐng)批件》及藥品批準(zhǔn)文號(hào)��。
對(duì)于進(jìn)口藥品�,制藥廠商名稱、注冊(cè)地址變更���,屬報(bào)“國家食品藥品監(jiān)督管理局審批的補(bǔ)充申請(qǐng)事項(xiàng)”��,無需進(jìn)行技術(shù)審評(píng)��。
改變國內(nèi)藥品生產(chǎn)企業(yè)名稱����,屬報(bào)“省級(jí)食品藥品監(jiān)督管理部門批準(zhǔn)國家食品藥品監(jiān)督管理局備案申請(qǐng)事項(xiàng)”���。
新法規(guī)環(huán)境
新修訂《藥品管理法》第四十條規(guī)定�����,“經(jīng)國務(wù)院藥品監(jiān)督管理部門批準(zhǔn)�����,藥品上市許可持有人可以轉(zhuǎn)讓藥品上市許可�����。受讓方應(yīng)當(dāng)具備保障藥品安全性��、有效性和質(zhì)量可控性的質(zhì)量管理����、風(fēng)險(xiǎn)防控和責(zé)任賠償?shù)饶芰?����,履行藥品上市許可持有人義務(wù)”��。新修訂《藥品注冊(cè)管理辦法》第七十八條將“持有人轉(zhuǎn)讓藥品上市許可”列為以補(bǔ)充申請(qǐng)方式申報(bào)的變更�。
《辦法》第二章第一節(jié)明確了持有人變更管理具體要求。
申請(qǐng)變更境內(nèi)生產(chǎn)藥品的持有人�����,受讓方應(yīng)當(dāng)在取得相應(yīng)生產(chǎn)范圍的藥品生產(chǎn)許可證后,向藥審中心提出補(bǔ)充申請(qǐng)����;境外持有人之間變更的,由變更后持有人向藥審中心提出補(bǔ)充申請(qǐng)��。藥審中心同意變更的�����,核發(fā)藥品補(bǔ)充申請(qǐng)通知書����,藥品批準(zhǔn)文號(hào)和證書有效期不變。
持有人名稱�、生產(chǎn)企業(yè)名稱、生產(chǎn)地址名稱等變更���,應(yīng)當(dāng)完成藥品生產(chǎn)許可證相應(yīng)事項(xiàng)變更后�����,向所在地省級(jí)藥品監(jiān)管部門就藥品批準(zhǔn)證明文件相應(yīng)管理信息變更進(jìn)行備案���。境外生產(chǎn)藥品上述信息的變更向藥審中心提出備案�。
同時(shí)�����,《辦法》附件4中明確了藥品上市許可持有人變更的申報(bào)資料要求����,資料要求中除提交歷次批準(zhǔn)文件和證明性文件外��,還需要提交受讓方對(duì)擬轉(zhuǎn)讓藥品的生產(chǎn)場(chǎng)地���、處方���、生產(chǎn)工藝、質(zhì)量標(biāo)準(zhǔn)等應(yīng)當(dāng)與原藥品一致�����、不發(fā)生變更的承諾��。
關(guān)于已在境內(nèi)上市的境外生產(chǎn)藥品轉(zhuǎn)移至境內(nèi)生產(chǎn)的情形
原法規(guī)環(huán)境
《關(guān)于印發(fā)藥品技術(shù)轉(zhuǎn)讓注冊(cè)管理規(guī)定的通知》規(guī)定����,已獲得進(jìn)口藥品注冊(cè)證的品種�,其生產(chǎn)技術(shù)可以由原進(jìn)口藥品注冊(cè)申請(qǐng)人轉(zhuǎn)讓給境內(nèi)藥品生產(chǎn)企業(yè)���,按照補(bǔ)充申請(qǐng)的程序和規(guī)定報(bào)送有關(guān)資料和說明�����。
新法規(guī)環(huán)境
《辦法》第十條規(guī)定��,已在境內(nèi)上市的境外生產(chǎn)藥品轉(zhuǎn)移至境內(nèi)生產(chǎn)的���,應(yīng)當(dāng)由境內(nèi)申請(qǐng)人按照藥品上市注冊(cè)申請(qǐng)的要求和程序提出申請(qǐng),相關(guān)藥學(xué)���、非臨床研究和臨床研究資料(適用時(shí))可提交境外生產(chǎn)藥品的原注冊(cè)申報(bào)資料����,符合要求的可申請(qǐng)成為參比制劑�。
同時(shí),該條的實(shí)施設(shè)置了2年的過渡期����,境內(nèi)持有人可在2023年1月15日前繼續(xù)按照《關(guān)于印發(fā)藥品技術(shù)轉(zhuǎn)讓注冊(cè)管理規(guī)定的通知》要求開展研究并申報(bào)補(bǔ)充申請(qǐng)���。
關(guān)于生產(chǎn)場(chǎng)地變更
原法規(guī)環(huán)境
原《藥品注冊(cè)管理辦法》附件4中明確,“國內(nèi)藥品生產(chǎn)企業(yè)內(nèi)部改變藥品生產(chǎn)場(chǎng)地”為經(jīng)省級(jí)藥監(jiān)部門批準(zhǔn)國家藥監(jiān)部門備案的補(bǔ)充申請(qǐng)事項(xiàng)�,申請(qǐng)資料中應(yīng)當(dāng)提供變更后的《藥品生產(chǎn)許可證》。因此��,變更藥品生產(chǎn)場(chǎng)地需由申請(qǐng)人向省級(jí)藥監(jiān)部門分別提出變更《藥品生產(chǎn)許可證》和藥品注冊(cè)批準(zhǔn)證明文件申請(qǐng)���。
改變進(jìn)口藥品的產(chǎn)地為報(bào)國家藥監(jiān)部門審批的補(bǔ)充申請(qǐng)事項(xiàng)����。
新法規(guī)環(huán)境
《辦法》中優(yōu)化藥品生產(chǎn)場(chǎng)地變更的申報(bào)程序���。
《辦法》明確,藥品上市后發(fā)生藥品生產(chǎn)場(chǎng)地變更的�����,只需向省級(jí)藥品監(jiān)管部門提出變更《藥品生產(chǎn)許可證》申請(qǐng)���?���!掇k法》第十四條對(duì)具體程序進(jìn)行了細(xì)化明確,《藥品生產(chǎn)許可證》變更獲得批準(zhǔn)后����,由省級(jí)藥品監(jiān)管部門直接在變更系統(tǒng)中更新藥品注冊(cè)批準(zhǔn)證明文件及其附件上的藥品生產(chǎn)場(chǎng)地變更信息。生物制品變更中涉及需要向藥審中心提出補(bǔ)充申請(qǐng)事項(xiàng)的���,持有人按照本辦法提出補(bǔ)充申請(qǐng)�。
對(duì)于境外持有人變更藥品生產(chǎn)場(chǎng)地且變更后生產(chǎn)場(chǎng)地仍在境外的�,應(yīng)按照相關(guān)技術(shù)指導(dǎo)原則進(jìn)行研究、評(píng)估和必要的驗(yàn)證�,向藥審中心提出補(bǔ)充申請(qǐng)或備案。
關(guān)于原料藥變更
原法規(guī)環(huán)境
原《藥品注冊(cè)管理辦法》附件4����,“改變進(jìn)口藥品制劑所用原料藥的產(chǎn)地”為報(bào)國家藥監(jiān)部門備案的補(bǔ)充申請(qǐng)事項(xiàng);“改變國內(nèi)生產(chǎn)藥品制劑的原料藥產(chǎn)地”為報(bào)省級(jí)藥監(jiān)部門備案的補(bǔ)充申請(qǐng)事項(xiàng)�。
新法規(guī)環(huán)境
結(jié)合新修訂《藥品注冊(cè)管理辦法》明確建立的化學(xué)原料藥、輔料及直接接觸藥品的包裝材料和容器關(guān)聯(lián)審評(píng)審批制度�,《辦法》進(jìn)一步規(guī)定原料藥變更原則。
已經(jīng)通過審評(píng)審批的原料藥發(fā)生變更的�,原料藥登記人應(yīng)按照現(xiàn)行藥品注冊(cè)管理有關(guān)規(guī)定、藥品生產(chǎn)質(zhì)量管理規(guī)范����、技術(shù)指導(dǎo)原則確定變更管理類別后經(jīng)批準(zhǔn)�����、備案后實(shí)施或報(bào)告�,相關(guān)信息由登記人及時(shí)在登記平臺(tái)更新����。變更實(shí)施前,原料藥登記人應(yīng)將有關(guān)情況及時(shí)通知相關(guān)制劑持有人�����,便于制劑持有人開展后續(xù)工作����。
未通過審評(píng)審批,且尚未進(jìn)入審評(píng)程序的原料藥發(fā)生變更的���,原料藥登記人可以通過藥審中心網(wǎng)站登記平臺(tái)隨時(shí)更新相關(guān)資料。
此外�����,《化學(xué)原料藥�、藥用輔料及藥包材與藥品制劑關(guān)聯(lián)審評(píng)審批管理規(guī)定(征求意見稿)》也明確�����,化學(xué)原料藥發(fā)生變更應(yīng)按照現(xiàn)行注冊(cè)管理有關(guān)規(guī)定執(zhí)行��,并及時(shí)告知關(guān)聯(lián)制劑上市許可持有人���,藥品上市許可持有人應(yīng)充分評(píng)估對(duì)制劑產(chǎn)品質(zhì)量的影響,并按相關(guān)管理規(guī)定進(jìn)行注冊(cè)申報(bào)��?!兑焉鲜谢瘜W(xué)藥品藥學(xué)變更研究技術(shù)指導(dǎo)原則(征求意見稿)》中列舉了變更原料藥生產(chǎn)工藝、變更原料藥生產(chǎn)場(chǎng)地和變更制劑所用原料藥的供應(yīng)商的風(fēng)險(xiǎn)分類適用情形�����。
其他注冊(cè)管理事項(xiàng)變更
原法規(guī)環(huán)境
原《藥品注冊(cè)管理辦法》附件4中�,將生產(chǎn)工藝、藥用輔料��、質(zhì)量標(biāo)準(zhǔn)等生產(chǎn)過程變更�����,按照監(jiān)管要求分為國家藥監(jiān)部門審批��、省級(jí)藥監(jiān)部門審批、國家藥監(jiān)部門備案�����、省級(jí)藥監(jiān)部門備案等四種情形�����。
新法規(guī)環(huán)境
《辦法》第十七條明確:生產(chǎn)設(shè)備�����、原輔料及包材來源和種類�、生產(chǎn)環(huán)節(jié)技術(shù)參數(shù)、質(zhì)量標(biāo)準(zhǔn)等生產(chǎn)過程變更的�,持有人應(yīng)當(dāng)充分評(píng)估該變更可能對(duì)藥品安全性、有效性和質(zhì)量可控性影響的風(fēng)險(xiǎn)程度��,確定變更管理類別���,按照有關(guān)技術(shù)指導(dǎo)原則和藥品生產(chǎn)質(zhì)量管理規(guī)范進(jìn)行充分研究����、評(píng)估和必要的驗(yàn)證��,經(jīng)批準(zhǔn)���、備案后實(shí)施或報(bào)告����。
持有人可依據(jù)藥審中心發(fā)布的《已上市化學(xué)藥品藥學(xué)變更研究技術(shù)指導(dǎo)原則(征求意見稿)》《已上市生物制品藥學(xué)變更研究技術(shù)指導(dǎo)原則(征求意見稿)》《已上市中藥藥學(xué)變更研究技術(shù)指導(dǎo)原則(征求意見稿)》等對(duì)相應(yīng)類別藥品的上市后變更進(jìn)行研究和分類��,也可自主選擇使用先進(jìn)的變更管理工具進(jìn)行管理��,按照風(fēng)險(xiǎn)分為重大變更����、中等變更和微小變更。對(duì)于無法確定變更管理類別的�,《辦法》第三章“變更管理類別確認(rèn)及調(diào)整”中也明確了與省級(jí)藥品監(jiān)管部門或藥審中心溝通及具體實(shí)施情況。
關(guān)于審批類變更獲得批準(zhǔn)后執(zhí)行時(shí)間問題
原法規(guī)環(huán)境
未予明確審批類變更獲得批準(zhǔn)后�,在藥品生產(chǎn)中何時(shí)實(shí)施。
新法規(guī)環(huán)境
《辦法》第二十五條明確:持有人應(yīng)當(dāng)在提出變更的補(bǔ)充申請(qǐng)時(shí)承諾變更獲得批準(zhǔn)后的實(shí)施時(shí)間�����,實(shí)施時(shí)間原則上最長不得超過自變更獲批之日起6個(gè)月����,涉及藥品安全性變更的事項(xiàng)除外����。
關(guān)于審評(píng)/備案時(shí)限問題
原法規(guī)環(huán)境
原《藥品注冊(cè)管理辦法》中明確了需要進(jìn)行技術(shù)審評(píng)的補(bǔ)充申請(qǐng)時(shí)限為40日���,對(duì)于合并申報(bào)事項(xiàng)及備案類補(bǔ)充申請(qǐng)時(shí)限未予明確���。
新法規(guī)環(huán)境
新修訂《藥品注冊(cè)管理辦法》中明確,審批類變更的補(bǔ)充申請(qǐng)審評(píng)時(shí)限為60日�,補(bǔ)充申請(qǐng)合并申報(bào)事項(xiàng)的,審評(píng)時(shí)限為80日�����,其中�����,涉及臨床試驗(yàn)研究數(shù)據(jù)審查��、藥品注冊(cè)核查檢驗(yàn)的審評(píng)時(shí)限為200日�。《辦法》中對(duì)備案事項(xiàng)的時(shí)限作出具體明確���,要求藥審中心或省級(jí)藥品監(jiān)管部門應(yīng)當(dāng)自備案完成之日起5日內(nèi)公示有關(guān)信息�����,省級(jí)藥品監(jiān)管部門自備案完成之日起30日內(nèi)完成對(duì)備案資料的審查�����,必要時(shí)可實(shí)施檢查與檢驗(yàn)�����。
(來源:中國醫(yī)藥報(bào))